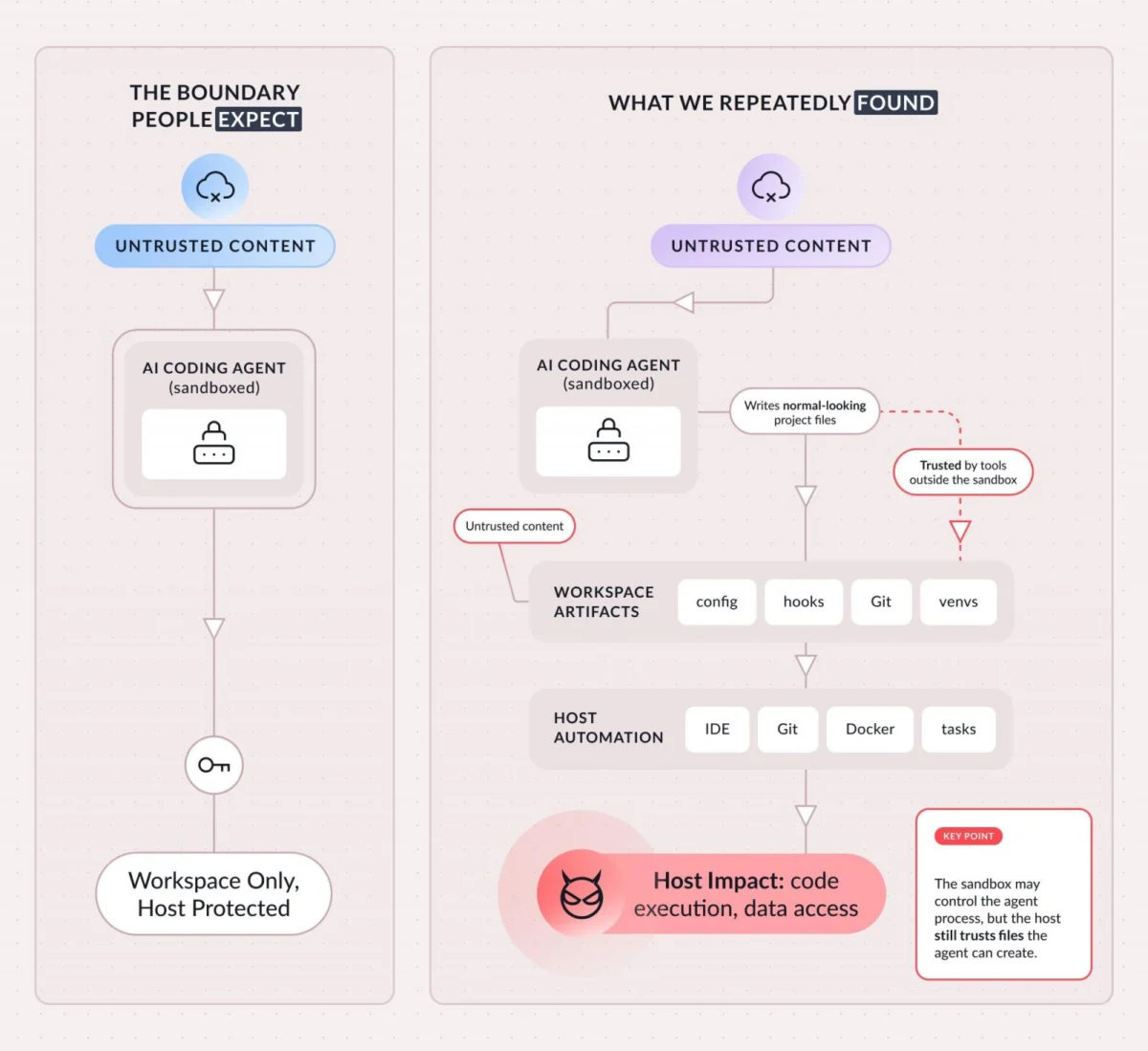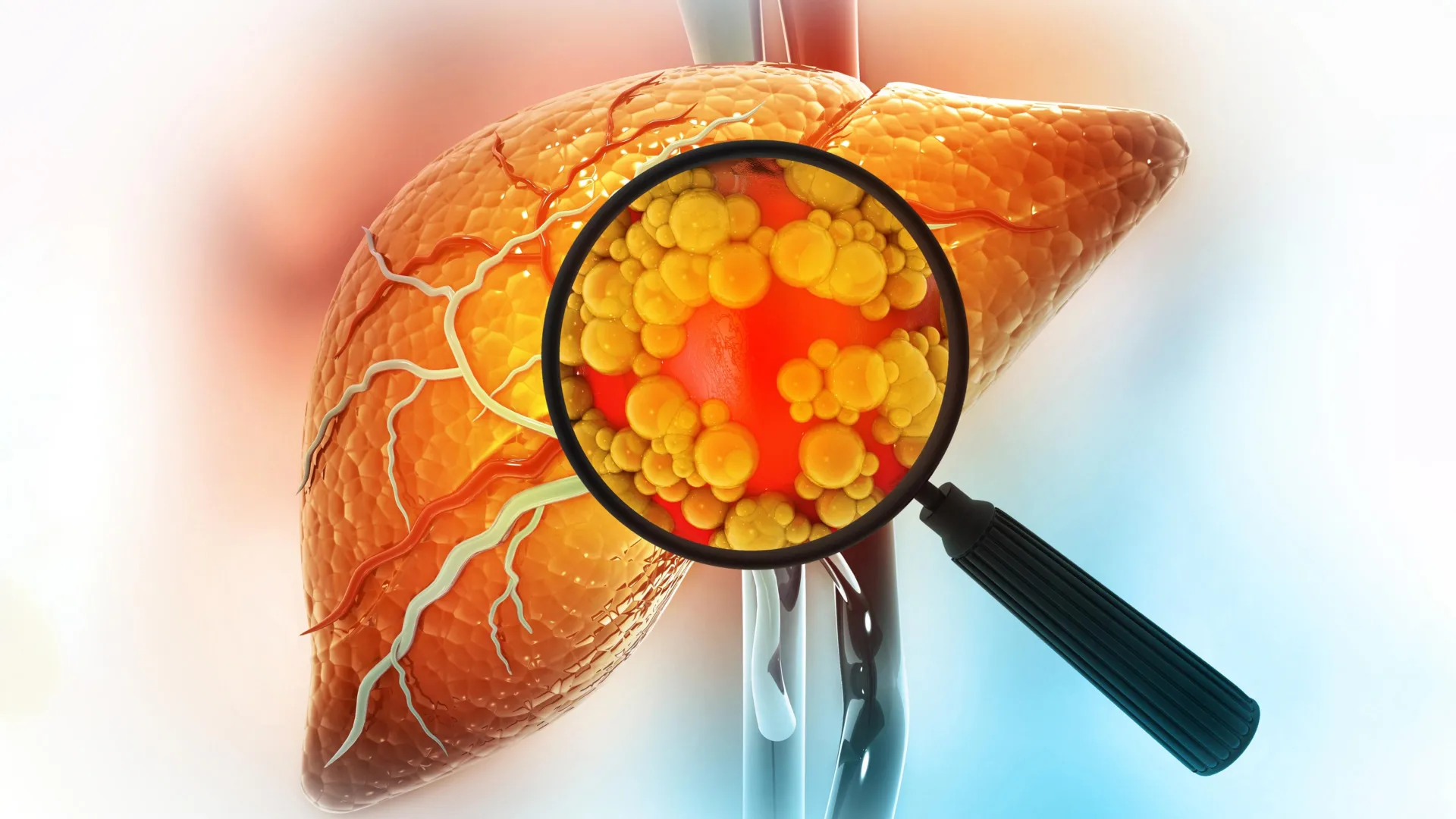
A recent groundbreaking investigation has illuminated a previously unrecognized mechanism contributing to chronic liver pathologies and systemic aging. This pivotal research demonstrates that the selective elimination of specific dysfunctional immune cells, known as senescent macrophages, can significantly ameliorate hepatic damage and inflammation in preclinical models, even in the persistence of adverse dietary conditions. These findings offer a profound new perspective on the etiology of age-related diseases and present a compelling therapeutic avenue for conditions such as non-alcoholic fatty liver disease (NAFLD) and potentially broader age-associated dysfunctions.
The study centers on the phenomenon of cellular senescence, a state in which cells, subjected to various stressors, cease their replicative activity but resist programmed cell death (apoptosis). Instead of being cleared, these lingering cells, colloquially termed "zombie cells," remain metabolically active within tissues. Critically, they adopt a detrimental secretory profile, known as the Senescence-Associated Secretory Phenotype (SASP), releasing a persistent cocktail of pro-inflammatory cytokines, chemokines, growth factors, and proteases. This continuous molecular barrage can disrupt local tissue homeostasis, induce senescence in neighboring healthy cells, and drive chronic, low-grade inflammation, which is a hallmark of aging and numerous age-related diseases. The accumulation of even a small percentage of these dysfunctional cells can exert disproportionately large negative effects on tissue function, akin to a persistent, insidious contaminant gradually degrading a complex system.
For an extended period, the precise role of macrophages, the body’s vigilant immune sentinels responsible for waste clearance and pathogen defense, in the context of cellular senescence remained a subject of considerable debate. Many researchers harbored skepticism regarding the capacity of macrophages to truly enter a senescent state. This uncertainty stemmed from the inherent complexity of macrophage biology; healthy, activated macrophages naturally exhibit some molecular characteristics that overlap with those typically observed in senescent cells, making definitive differentiation a significant challenge. The ambiguity created a critical void in understanding their potential contribution to age-related tissue dysfunction.
Addressing this diagnostic ambiguity, the research team achieved a significant breakthrough by identifying a robust and unambiguous molecular signature for senescent macrophages. Their meticulous analysis revealed that the co-expression of two specific proteins, p21 and TREM2, reliably serves as a biomarker for macrophages that have not only entered senescence but are also actively contributing to inflammation and tissue damage, rather than performing their beneficial immune functions. P21, a cyclin-dependent kinase inhibitor, is a well-established marker of cell cycle arrest, while TREM2 (Triggering Receptor Expressed on Myeloid cells 2) is a receptor often associated with immune cell activation, phagocytosis, and inflammation resolution. Its aberrant expression in conjunction with p21 indicates a dysfunctional, pro-inflammatory state in these senescent immune cells.
Armed with this novel molecular marker, the investigators were able to quantify the accumulation of senescent macrophages across different age cohorts. Their observations in murine models revealed a stark and concerning age-dependent increase. In youthful mice, only approximately 5% of liver-resident macrophages exhibited the p21-TREM2 signature. However, in older animals, this proportion surged dramatically, reaching between 60% and 80%. This profound age-related escalation in senescent macrophages directly correlated with a parallel increase in chronic hepatic inflammation, providing compelling evidence for their causal involvement in age-related liver deterioration.
Beyond chronological aging, the research uncovered another critical trigger for macrophage senescence: sustained exposure to excessive cholesterol. The investigators demonstrated that healthy macrophages, when cultured in vitro and exposed to high concentrations of low-density lipoprotein (LDL) cholesterol – often termed "bad cholesterol" – underwent a transformation into a senescent state. These cells ceased division, began secreting pro-inflammatory proteins characteristic of the SASP, and prominently displayed the p21-TREM2 signature. This mechanistic link highlights how chronic metabolic overload, particularly from dietary sources, can directly induce cellular senescence in immune cells.
Under normal physiological conditions, macrophages are adept at metabolizing and clearing cholesterol, playing a vital role in lipid homeostasis. However, the study suggests that chronic and excessive exposure to cholesterol, as is often seen in conditions of overnutrition and dyslipidemia, overwhelms these metabolic pathways, pushing macrophages into a pathological, senescent state. This finding is particularly pertinent to the understanding of fatty liver disease, a condition strongly associated with high-fat, high-cholesterol diets. The accumulation of excess cholesterol in the bloodstream and liver tissue appears to be a primary driver for the expansion of this detrimental senescent macrophage population.
The implications of this discovery extend beyond the liver. The identification of cholesterol as a potent inducer of macrophage senescence raises the significant possibility that diets rich in saturated fats and cholesterol may accelerate biological aging not only in the liver but also in a multitude of other vital organs. Macrophages and their tissue-specific counterparts, such as microglia in the brain, Kupffer cells in the liver, and adipose tissue macrophages, are ubiquitous throughout the body. Their widespread senescence, triggered by dietary factors, could therefore contribute to a broader spectrum of age-related pathologies, including atherosclerosis, neurodegenerative disorders, and metabolic dysregulation in adipose tissue.
To definitively test the therapeutic potential of removing these senescent cells, the research team administered ABT-263, a pharmacological agent known as a senolytic drug, specifically designed to induce apoptosis in senescent cells, to the murine models. The outcomes of this intervention were remarkably compelling. In mice that had been maintained on a high-fat, high-cholesterol diet – a regimen designed to induce fatty liver disease – treatment with ABT-263 led to a dramatic reduction in liver size, from an average of approximately 7% of total body weight to a significantly healthier range of 4-5%. Concurrently, the overall body weight of the treated animals decreased by roughly 25%, falling from approximately 40 grams to around 30 grams.
Visual inspection further underscored the efficacy of the treatment. The livers of the ABT-263-treated mice appeared considerably smaller, exhibited a normal, healthy red coloration, and displayed a normalized architecture, a stark contrast to the enlarged, visibly yellowish, and structurally compromised livers observed in the untreated control group. These results robustly suggest that the targeted elimination of senescent macrophages alone is sufficient to induce substantial metabolic improvements and reverse significant liver damage, critically, even in the absence of any dietary modifications. This independence from dietary change was a particularly striking finding, indicating that clearing these dysfunctional cells can effectively interrupt the disease progression regardless of the initial dietary insult. The reversal of fatty liver disease, rather than merely slowing its progression, represents a profound therapeutic paradigm shift.
Translational relevance to human health was a key consideration. To ascertain whether these findings might apply to human chronic liver disease, the researchers meticulously analyzed existing genomic datasets derived from human liver biopsies. This analysis confirmed that the identical p21-TREM2 senescent macrophage signature was significantly more prevalent and expressed at higher levels in diseased human livers compared to healthy controls. This robust correlation strongly suggests that macrophage senescence plays a contributing role in the pathogenesis of chronic liver disease in humans, mirroring the observations in the murine models.
The implications for public health are substantial, particularly in regions facing a high burden of metabolic diseases. In metropolitan areas like Los Angeles, for instance, an estimated 30-40% of the population is affected by fatty liver disease, with disproportionately higher rates observed within specific ethnic communities, such as Latino populations. The current treatment landscape for fatty liver disease remains remarkably limited, often relying on lifestyle modifications that can be challenging to sustain, and early detection tools are still largely inadequate. This burgeoning epidemic of fatty liver disease, affecting progressively younger demographics, underscores the urgent need for novel therapeutic strategies.
While the senolytic agent ABT-263 proved highly effective in the preclinical mouse models, its systemic toxicity precludes widespread therapeutic use in humans. Known side effects, including bone marrow suppression and gastrointestinal distress, render it unsuitable for chronic administration. Consequently, a primary objective for the research team involves an extensive screening effort to identify safer, more selective compounds that can specifically target and eliminate senescent macrophages without eliciting harmful off-target effects in human patients. This next phase of drug discovery is critical for translating these foundational insights into viable clinical interventions.
Furthermore, the investigators are actively exploring whether similar mechanisms of macrophage senescence contribute to other age-related pathologies. In the central nervous system, for example, microglia – the brain’s resident macrophages – are known to become dysfunctional in conditions such as Alzheimer’s disease, where they encounter and attempt to clear vast amounts of cellular debris and amyloid plaques. It is hypothesized that chronic exposure to these stressors could induce microglial senescence, thereby exacerbating neuroinflammation and neurodegeneration. Understanding this shared etiology across different organ systems could unlock broad therapeutic potential.
These findings lend compelling support to the geroscience hypothesis, an emerging paradigm that posits that targeting fundamental biological processes of aging can simultaneously prevent or treat a wide array of age-related diseases, rather than addressing each condition in isolation. In this specific context, the accumulation of senescent macrophages, driven by factors such as aging and chronic metabolic stress, emerges as a fundamental mechanism that could contribute to the pathogenesis of diverse conditions, ranging from fatty liver disease and atherosclerosis to Alzheimer’s disease and various forms of cancer. By deciphering and intervening in the basic cellular mechanisms that lead to the emergence of these dysfunctional cells, researchers aim to develop transformative therapies that not only mitigate specific diseases but also modulate the broader trajectory of human aging and health span.
This comprehensive study, supported by prestigious institutions including the National Institutes of Health, the Glenn Foundation for Medical Research, the American Federation for Aging Research, and the UCLA-UCSD Diabetes Research Center, represents a significant advancement in the understanding of age-related diseases and offers a beacon of hope for developing effective interventions. The ability to reverse significant organ damage by targeting a specific population of "zombie" cells marks a profound step forward in precision medicine and the burgeoning field of geroscience, holding the promise of healthier aging for a global population.